Binning and Functional Annotation
In this lesson we will finish off our analysis by separating out the individual genomes into metagenome-assembled genomes (MAGs) using a process called binning. Binning can be done in lots of different ways but the general idea is to put all the “similar” contigs together into one bin. By the end we should have several bins each containing the genome of one organism - this is what we call a MAG.
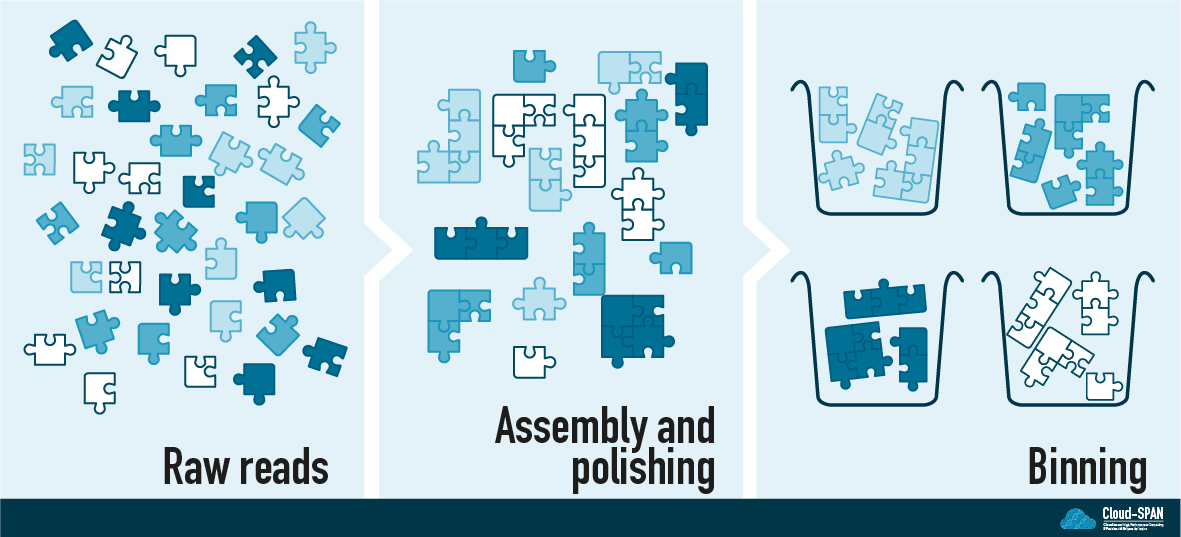
Once we have our MAGs we can annotate them to predict where genes are and what function they have. This is useful for predicting the metabolic capacity of each organism. We can also look up these predicted gene sequences in a database and build taxonomic trees to make an educated guess about what species we are looking at.
By the end of this lesson you will be able to:
- prepare your polished assembly for binning using BWA and Samtools
- generate bins using MetaBAT2
- evaluate the quality of your generated bins/MAGs using CheckM
- use Prokka for functional annotation of your MAGs