Redirection
Searching files
We discussed in a previous episode how to search within a file using less. We can also search within files without even opening them, using grep. grep is a command-line utility for searching plain-text files for lines matching a specific set of characters (sometimes called a string) or a particular pattern (which can be specified using something called regular expressions). We’re not going to work with regular expressions in this lesson, and are instead going to specify the strings we are searching for. Let’s give it a try!
The four nucleotides that appear in DNA are abbreviated A, C, T and G. Unknown nucleotides are represented with the letter N. An N appearing in a sequencing file represents a position where the sequencing machine was not able to confidently determine the nucleotide in that position. You can think of an N as being aNy nucleotide at that position in the DNA sequence.
We’ll search for strings inside of our fastq files. Let’s first make sure we are in the correct directory:
Code
cd ~/cs_course/data/raw_readsHowever, suppose we want to see how many reads in our file have bad segments containing three or more unclassified nucleotides (N) in a row.
In this lesson, we’re going to be manually searching for strings of bases within our sequence results to illustrate some principles of file searching. It can be really useful to do this type of searching to get a feel for the quality of your sequencing results, however, in your research you will most likely use a bioinformatics tool that has a built-in program for filtering out low-quality reads. You’ll learn how to use one such tool in a later lesson.
To search files we use a new command called grep. The name “grep” comes from an abbreviation of global regular expression print.
Let’s search for the string NN in the ERR4998593_1 file:
Code
grep NN SRR6820491_1.fastqThis command returns quite a lot of output to the terminal. Every single line in the SRR6820491_1 file that contains at least 2 consecutive Ns is printed to the terminal, regardless of how long or short the file is.
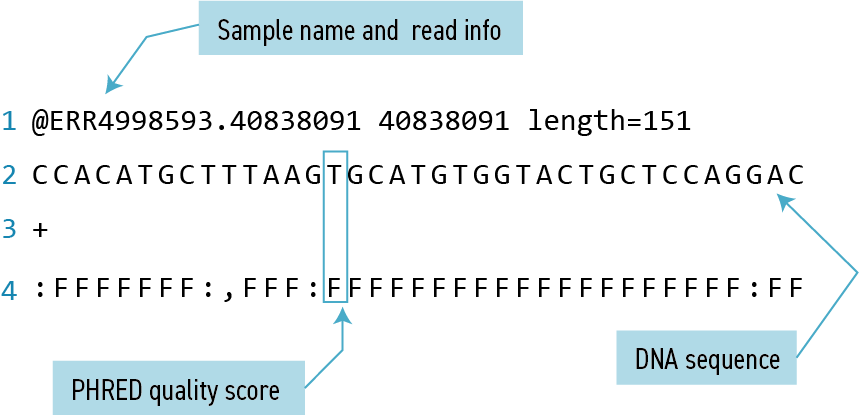
We may be interested not only in the actual sequence which contains this string, but in the name (or identifier) of that sequence. Think back to the FASTQ format we discussed previously - if you need a reminder, you can click to reveal one below.
To get all of the information about each read, we will return the line immediately before each match and the two lines immediately after each match.
We can use the -B argument for grep to return a specific number of lines before each match. The -A argument returns a specific number of lines after each matching line. Here we want the line before and the two lines after each matching line, so we add -B1 -A2 to our grep command:
Code
grep -B1 -A2 NN SRR6820491_1.fastqOne of the sets of lines returned by this command is:
Output
@SRR6820491.2176700 2176700 length=151
CCCGNAGATAGCTGGTTCTCCCCGAAATAGCTTTANGGCTANCCTCAGGTTAGTCNATCGGAGGTAAAGCACTGACTGGACTAGGGGCCTTANCGGGTTACCGAATCCTATCANACTCNNAATGCNGAATAGNTGATGCCTGGGAGNCAGA
+SRR6820491.2176700 2176700 length=151
AAFF#JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ#JJJJJ#JJJJJJJJJJJJJ#JJJJJFJJFJJJJJJJJJJJAJJJJJJJJJJJJJJJ#JJJJJJJJJJJJJJJJJJJJ#JJJJ##JJJJJ#-FFAFJ#FJFFJJJJJJJ-F#F-FASearch for the sequence
GGGCGCGTAGGCGin theSRR6820491_1.fastqfile. Have your search return all matching lines and the name (or identifier) for each sequence that contains a match. What is the output?Search for the sequence
GGGCGCGTAGGCGin both FASTQ files. Have your search return all matching lines and the name (or identifier) for each sequence that contains a match. How many matching sequences do you get? (this may take up to 4 minutes to complete so be patient!)
grep -B1 GGGCGCGTAGGCG SRR6820491_1.fastq
Output
grep -B1 GGGCGCGTAGGCG *.fastq
Output
Redirecting output
grep allowed us to identify sequences in our FASTQ files that match a particular pattern. All of these sequences were printed to our terminal screen, but in order to work with these sequences and perform other operations on them, we will need to capture that output in some way.
We can do this with something called “redirection”. The idea is that we are taking what would ordinarily be printed to the terminal screen and redirecting it to another location. In our case, we want to print this information to a file so that we can look at it later and use other commands to analyse this data.
Redirecting output to a file — the > command
The command for redirecting output to a file is >.
Let’s try out this command and copy all the records (including all four lines of each record) in our FASTQ files that contain ‘NN’ to another file called bad_reads.txt. The new flag --no-group-separator stops grep from putting a dashed line (–) between matches. The reason this is necessary will become apparent shortly.
Code
grep -B1 -A2 NN --no-group-separator SRR6820491_1.fastq > bad_reads.txtThe prompt should sit there a little bit, and then it should look like nothing happened. But type ls. You should see a new file called bad_reads.txt.
You might be confused about why we’re naming our output file with a .txt extension. After all, it will be holding FASTQ formatted data that we’re extracting from our FASTQ files.
Won’t it also be a FASTQ file?
The answer is, yes - it will be a FASTQ file and it would make sense to name it with a .fastq extension.
However, using an extension such as .txt makes it easy to distinguish the files you may generate through some exploratory processing, as the one we just made with the grep program, from the original sequencing files of your project. So you can easily select all the files with a specific extension for further processing using the wildcard * character, for example: grep .. *.fastq or mv *.txt newlocation.
Counting number of lines in files — the wc program
We can check the number of lines in our new file using a command called wc. wc stands for word count. This command counts the number of words, lines, and characters in a file.
Code
wc bad_reads.txtOutput
208 416 19864 bad_reads.txtThis will tell us the number of lines, words and characters in the file. If we want only the number of lines, we can use the -l flag for lines.
Code
wc -l bad_reads.txtOutput
208 bad_reads.txtThe --no-group-separator flag used above prevents grep from adding unnecessary extra lines to the file which would alter the number of lines present.
- How many sequences are there in
SRR6820491_1.fastq? Remember that every sequence is formed by four lines. - How many sequences in
SRR6820491_1.fastqcontain at least 5 consecutive Ns?
Code
wc -l SRR6820491_1.fastqOutput
56397388 SRR6820491_1.fastqNow you can divide this number by four to get the number of sequences in your fastq file (34263610).
Code
grep NNNNN SRR6820491_1.fastq > bad_reads.txt
wc -l bad_reads.txtOutput
0 bad_reads.txtThe command > redirects and overwrites
We might want to search multiple FASTQ files for sequences that match our search pattern. However, we need to be careful, because each time we use the > command to redirect output to a file, the new output will replace the output that was already present in the file. This is called “overwriting” and, just like you don’t want to overwrite your video recording of your kid’s first birthday party, you also want to avoid overwriting your data files.
Code
grep -B1 -A2 NN --no-group-separator SRR6820491_1.fastq > bad_reads.txt
wc -l bad_reads.txtOutput
208 bad_reads.txtCode
grep -B1 -A2 NNNNNNNNNN --no-group-separator SRR6820491_1.fastq > bad_reads.txt
wc -l bad_reads.txtOutput
0 bad_reads.txtHere, the output of our second call to wc shows that we no longer have any lines in our bad_reads.txt file. This is because the second string we searched (NNNNNNNNNN) does not match any strings in the file. So our file was overwritten and is now empty.
The command >> redirects and appends
We can avoid overwriting our files by using the command >>. >> is known as the “append redirect” and will append new output to the end of a file, rather than overwriting it.
Code
grep -B1 -A2 NN --no-group-separator SRR6820491_1.fastq > bad_reads.txt
wc -l bad_reads.txtOutput
208 bad_reads.txtCode
grep -B1 -A2 NNNNNNNNNN --no-group-separator SRR6820491_1.fastq >> bad_reads.txt
wc -l bad_reads.txtOutput
208 bad_reads.txtThe output of our second call to wc shows that we have not overwritten our original data.
Redirecting output as input to other program — the pipe | command
Since we might have multiple different criteria we want to search for, creating a new output file each time has the potential to clutter up our workspace. We also thus far haven’t been interested in the actual contents of those files, only in the number of reads that we’ve found. We created the files to store the reads and then counted the lines in the file to see how many reads matched our criteria. There’s a way to do this, however, that doesn’t require us to create these intermediate files - the pipe command (|).
This is probably not a key on your keyboard you use very much, so let’s all take a minute to find that key. For the standard QWERTY keyboard layout, the | character can be found using the key combination
- Shift+\
What | does is take the output that is scrolling by on the terminal and uses that output as input to another command. When our output was scrolling by, we might have wished we could slow it down and look at it, like we can with less. Well it turns out that we can! We can redirect our output from our grep call through the less command.
Code
grep -B1 -A2 NN SRR6820491_1.fastq | lessWe can now see the output from our grep call within the less interface. We can use the up and down arrows to scroll through the output and use q to exit less.
If we don’t want to create a file before counting lines of output from our grep search, we could directly pipe the output of the grep search to the command wc -l. This can be helpful for investigating your output if you are not sure you would like to save it to a file.
Code
grep -B1 -A2 NN --no-group-separator SRR6820491_1.fastq | wc -lOutput
208grep control
Use man grep to read more about other options to customize the output of grep including extended options, anchoring characters, and much more.
Redirecting output is often not intuitive, and can take some time to get used to. Once you’re comfortable with redirection, however, you’ll be able to combine any number of commands to do all sorts of exciting things with your data!
We’ll be using the redirect > and pipe | later in the course as part of our analysis workflow, so you will get lots of practice using them.
None of the command line programs we’ve been learning do anything all that impressive on their own, but when you start chaining them together, you can do some really powerful things very efficiently.