Project organisation
Overview
Teaching: 15 min
Exercises: 15 minQuestions
How can I organise my file system for a new bioinformatics project?
How can I document my work?
Objectives
Create a file system for a bioinformatics project.
Explain what types of files should go in your
docs,data, andresultsdirectories.Use the
historycommand and a text editor likenanoto document your work on your project.
Getting your project started
Project organisation is one of the most important parts of a sequencing project, and yet is often overlooked amidst the excitement of getting a first look at new data. Of course, while it’s best to get yourself organised before you even begin your analyses, it’s never too late to start, either.
You should approach your sequencing project similarly to how you do a biological experiment and this ideally begins with experimental design. We’re going to assume that you’ve already designed a beautiful sequencing experiment to address your biological question, collected appropriate samples, and that you have enough statistical power to answer the questions you’re interested in asking. These steps are all incredibly important, but beyond the scope of our course. For all of those steps (collecting specimens, extracting DNA, prepping your samples) you’ve likely kept a lab notebook that details how and why you did each step. However, the process of documentation doesn’t stop at the sequencer!
Genomics projects can quickly accumulate hundreds of files across tens of folders. Every computational analysis you perform over the course of your project is going to create many files, which can especially become a problem when you’ll inevitably want to run some of those analyses again. For instance, you might have made significant headway into your project, but then have to remember the PCR conditions you used to create your sequencing library months prior.
Other questions might arise along the way:
- What were your best alignment results?
- Which folder were they in: Analysis1, AnalysisRedone, or AnalysisRedone2?
- Which quality cutoff did you use?
- What version of a given program did you implement your analysis in?
Good documentation is key to avoiding this issue, and luckily enough, recording your computational experiments is even easier than recording lab data. Copy/Paste will become your best friend, sensible file names will make your analysis understandable by you and your collaborators, and writing the methods section for your next paper will be easy! Remember that in any given project of yours, it’s worthwhile to consider a future version of yourself as an entirely separate collaborator. The better your documentation is, the more this ‘collaborator’ will feel indebted to you!
With this in mind, let’s have a look at the best practices for documenting your genomics project. Your future self will thank you.
In this exercise we will setup a file system for the project we will be working on during this workshop.
We will start by creating a directory that we can use for the rest of the workshop. First navigate to your home directory. Then confirm that you are in the correct directory using the pwd command.
$ cd
$ pwd
You should see the output:
/home/csuser
Tip
If you aren’t in your home directory, the easiest way to get there is to enter the command
cd, which always returns you to home.
Exercise
Use the
mkdircommand to make the following directories:
cs_coursecs_course/docscs_course/datacs_course/resultsSolution
$ mkdir cs_course $ mkdir cs_course/docs $ mkdir cs_course/data $ mkdir cs_course/results
Use ls -R to verify that you have created these directories. The -R option for ls stands for recursive. This option causes
ls to return the contents of each subdirectory within the directory
iteratively.
$ ls -R cs_course
You should see the following output:
cs_course/:
data docs results
cs_course/data:
cs_course/docs:
cs_course/results:
If we were to draw this new file structure as a hierarchy tree, here’s what it would look like:
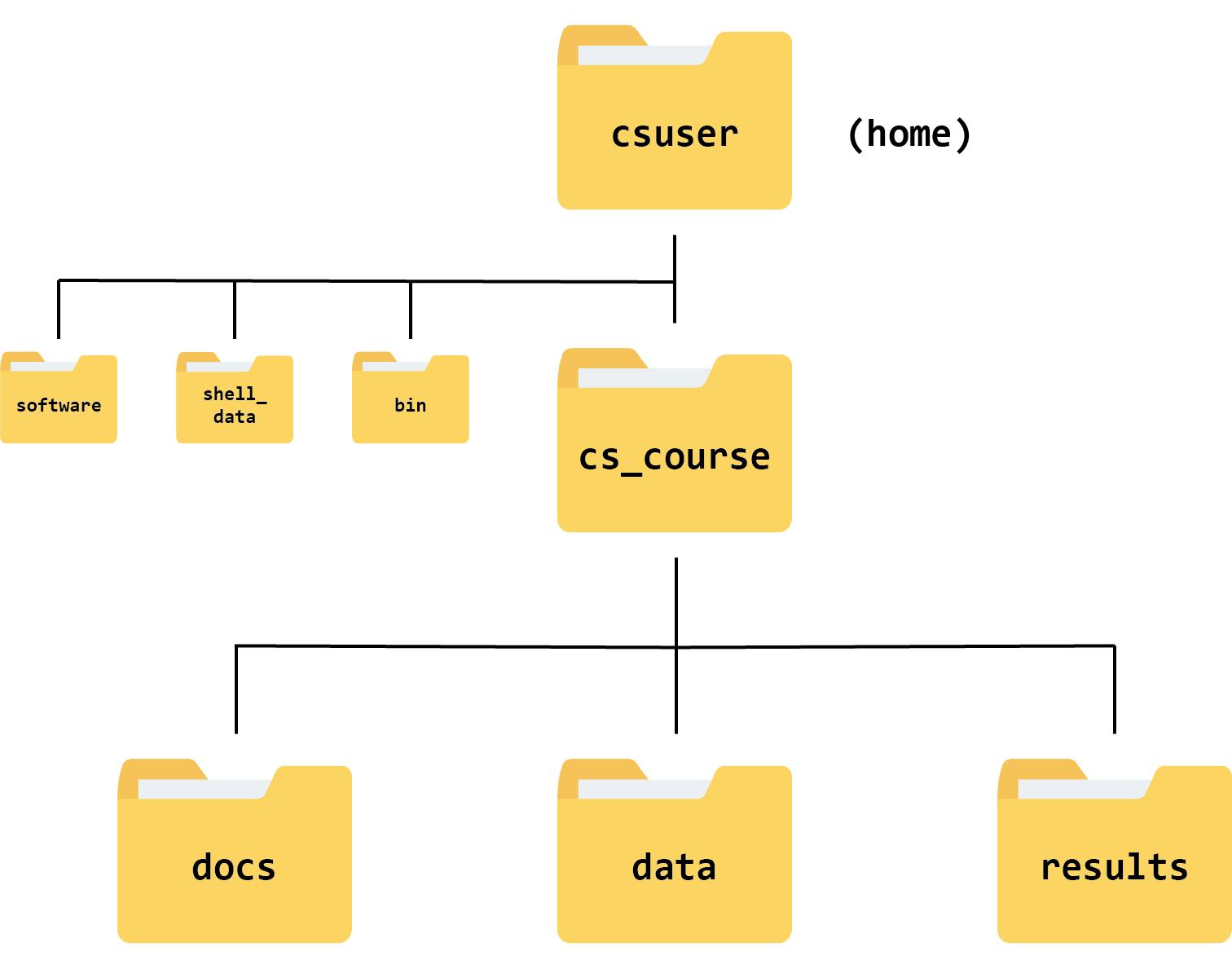
Organising your files
Before beginning any analysis, it’s important to save a copy of your raw data. The raw data should never be changed. Regardless of how sure you are that you want to carry out a particular data cleaning step, there’s always the chance that you’ll change your mind later or that there will be an error in carrying out the data cleaning and you’ll need to go back a step in the process. Having a raw copy of your data that you never modify guarantees that you will always be able to start over if something goes wrong with your analysis. When starting any analysis, you can make a copy of your raw data file and do your manipulations on that file, rather than the raw version. We learned in a previous episode how to prevent overwriting our raw data files by setting restrictive file permissions.
You can store any results that are generated from your analysis in
the results folder. This guarantees that you won’t confuse results
file and data files in six months or two years when you are looking
back through your files in preparation for publishing your study.
The docs folder is the place to store any written analysis of your
results, notes about how your analyses were carried out, and
documents related to your eventual publication.
Documenting your activity on the project
When carrying out wet-lab analyses, most scientists work from a written protocol and keep a hard copy of written notes in their lab notebook, including any things they did differently from the written protocol. This detailed record-keeping process is just as important when doing computational analyses. Luckily, it’s even easier to record the steps you’ve carried out computational than it is when working at the bench.
The history command is a convenient way to document all the
commands you have used while analysing and manipulating your project
files. Let’s document the work we have done on our project so far.
View the commands that you have used so far during this session using history:
$ history
The history likely contains many more commands than you have used for the current project. Let’s view the last several commands that focus on just what we need for this project.
View the last n lines of your history (where n = approximately the last few lines you think relevant). For our example, we will use the last 7:
$ history | tail -n 7
Exercise
Make sure you are in the
csuserdirectory.$ cdUsing your knowledge of the shell, use the append redirect
>>to create a file calledcs_course_log_XXXX_XX_XX.shwhich contains your last 7 commands. (Use the four-digit year, two-digit month, and two digit day, e.g.cs_course_log_2021_10_27.sh).Solution
$ history | tail -n 7 >> cs_course_log_2021_10_27.shNote we used the last 7 lines as an example, the number of lines may vary.
You may have noticed that your history contains the history command itself. To remove this redundancy
from our log, let’s use the nano text editor to fix the file:
$ nano cs_course_log_2021_10_27.sh
(Remember to replace the 2021_10_27 with your workshop date.)
From the nano screen, you can use your cursor to navigate, type, and delete any redundant lines.
Navigating in Nano
Although
nanois useful, it can be frustrating to edit documents, as you can’t use your mouse to navigate to the part of the document you would like to edit. Here are some useful keyboard shortcuts for moving around within a text document innano. You can find more information by typing Ctrl-G withinnano.
key action Ctrl-Space OR Ctrl-→ to move forward one word Alt-Space OR Esc-Space OR Ctrl-← to move back one word Ctrl-A to move to the beginning of the current line Ctrl-E to move to the end of the current line Ctrl-W to search
Add a date line and comment to the line where you have created the directory. Recall that any
text on a line after a # is ignored by bash when evaluating the text as code. For example:
# 2021_10_27
# Created sample directories for Cloud-SPAN workshop
Next, remove any lines of the history that are not relevant by navigating to those lines and using your delete key.
You will also need to delete the line numbers at the start of each line.
Save your file and close nano.
Your file should look something like this:
# 2021_10_27
# Created sample directories for Cloud-SPAN workshop
mkdir cs_course
mkdir cs_course/docs
mkdir cs_course/data
mkdir cs_course/results
If you keep this file up to date, you can use it to re-do your work on your project if something happens to your results files. To demonstrate how this works, first delete
your cs_course directory and all of its subdirectories. Look at your directory
contents to verify the directory is gone.
Make sure your log is not in the directory you’re about to delete! If it is, use mv [log-name] /home/csuser to move it into your home directory.
$ rm -r cs_course
$ ls
shell_data cs_course_log_2021_10_27.sh
Then run your workshop log file as a bash script. You should see the cs_course
directory and all of its subdirectories reappear.
$ bash cs_course_log_2021_10_27.sh
$ ls
shell_data cs_course cs_course_log_2021_10_27.sh
It’s important that we keep our workshop log file outside of our cs_course directory
if we want to use it to recreate our work. It’s also important for us to keep it up to
date by regularly updating with the commands that we used to generate our results files.
Congratulations! You’ve finished your introduction to writing scripts for genomics projects. You now know how to automate repetitive tasks using scripts and wildcards. With this solid foundation, you’re ready to move on to apply all of these new skills to carrying out more sophisticated bioinformatics analysis work. Don’t worry if everything doesn’t feel perfectly comfortable yet. We’re going to have many more opportunities for practice as we move forward on our bioinformatics journey!
References
A Quick Guide to organising Computational Biology Projects
Key Points
Spend the time to organise your file system when you start a new project. Your future self will thank you!
Always save a write-protected copy of your raw data.