Trimming and Filtering
Overview
Teaching: 30 min
Exercises: 25 minQuestions
How can I get rid of sequence data that doesn’t meet my quality standards?
Objectives
Clean FASTQ reads using cutadapt.
Select and set multiple options for command-line bioinformatic tools.
Write
forloops with two variables.
Cleaning Reads
In the previous episode, we took a high-level look at the quality of each of our samples using FastQC. We visualized per-base quality graphs showing the distribution of read quality at each base across all reads in a sample and extracted information about which samples fail which quality checks. Some of our samples failed quite a few quality metrics used by FastQC. This doesn’t mean, though, that our samples should be thrown out! It’s very common to have some quality metrics fail, and this may or may not be a problem for your downstream application. For our variant calling workflow, we will be removing some of the low quality sequences to reduce our false positive rate due to sequencing error.
We will use a program called cutadapt to filter poor quality reads and trim poor quality bases from our samples.
Cutadapt Options
Cutadapt has a variety of options to trim your reads. If we run the following command, we can see some of our options.
$ cutadapt
Which will give you the following output:
This is cutadapt 3.5 with Python 3.9.5
Command line parameters:
Run "cutadapt --help" to see command-line options.
See https://cutadapt.readthedocs.io/ for full documentation.
cutadapt: error: You did not provide any input file names. Please give me something to do!
In order to see the parameter options we need to pass -h as follows:
$ cutadapt -h
There are many parameters here so we will just show the top of the output which explains the usage, but you should look through and familiarise yourself with the options available. Importantly it shows you what are the required parameters and also the version of the software you have used which is important to keep note of. You should always record what versions of software you have used and what parameters you ran the software with in order to make your analysis reproducible
cutadapt version 3.5
Copyright (C) 2010-2021 Marcel Martin <marcel.martin@scilifelab.se>
cutadapt removes adapter sequences from high-throughput sequencing reads.
Usage:
cutadapt -a ADAPTER [options] [-o output.fastq] input.fastq
For paired-end reads:
cutadapt -a ADAPT1 -A ADAPT2 [options] -o out1.fastq -p out2.fastq in1.fastq in2.fastq
This output shows us that there is a different usage for single end or paired end reads. For single ended data we need to use the (-a) flag to specify the sequence of the adaptor to be removed, (-o) to specify the name of the trimmed output, and then the name of the raw sequence we want to be trimmed after. For paired end data, cutadapt expects two adaptor sequences for trimming, specified by (-a) and (-A). The output again is specified by (-o) for one trimmed read file, and (-p) for the other trimmed read file. The two files to be trimmed are then given as the last arguements.
The first input file typically contains a _1 or _R1 in the name
The second input file typically contains a _2 or _R2 in the name
For more information about the arguments used see the cutadapt documentation
An example command for cutadapt will look like the command below. This command is an example and will not work, as we do not have the files it refers to:
$ cutadapt -q 20 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCA \
-A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT \
-o trimmed.R1.fastq.gz -p trimmed.R2.fastq.gz \
reads.R1.fastq.gz reads.R2.fastq.gz \
> sample_fastq.gz.log &
In this example, we’ve told cutadapt:
| code | meaning |
|---|---|
-q 20 |
Bases need to have a quality phred score of at least 20 to be maintained |
-a |
The sequence to trim from the forward reads, this is based on illuminas truseq adaptors |
-A |
The sequence to trim from the reverse reads, this is also based on illumina truseq adaptors |
-o trimmed.R1.fastq.gz |
the name of the output of the trimmed forward reads |
-p trimmed.R2.fastq.gz |
the name of the output of the trimmed reverse reads |
reads.R1.fastq.gz |
the input forward reads file |
reads.R2.fastq.gz |
the input reverse reads file |
sample_fastq.gz.log |
this writes the output log of cutadapt to a file |
Multi-line commands
Some of the commands we ran in this lesson are long! When typing a long command into your terminal, you can use the
\character to separate code chunks onto separate lines. This can make your code more readable.
Running cutadapt
Now we will run cutadapt on our data. To begin, navigate to your untrimmed_fastq data directory:
$ cd ~/cs_course/data/untrimmed_fastq
We are going to run cutadapt on one of our paired-end samples. While using FastQC we saw that Nextera adapters were present in our samples. The adaptor sequences for both reads according to the illumina website is CTGTCTCTTATACACATCT
We will remove bases if their phred score is below 20 (like in our example above). The ampersand (&) tells linux to run the process in the background and the greater than symbol (>) pipes the output of the command to a file.
$ cutadapt -q 20 -a CTGTCTCTTATACACATCT \
-A CTGTCTCTTATACACATCT \
-o SRR2589044_1.trim.fastq.gz -p SRR2589044_2.trim.fastq.gz \
SRR2589044_1.fastq.gz SRR2589044_2.fastq.gz \
> SRR2589044_fastq.gz.log &
When you have submitted the command, you will see a number in square brackets like below, this is the number assigned to the job on the system so the number will vary. You will know when your job is done when the output says Done
[1] 17348
Done 00:01:10 1,107,090 reads @ 63.3 µs/read; 0.95 M reads/minute
The program will take around one minute to run. You will need to press enter to exit once it has finished.
We told the computer to run the process in the background (using &) so to see our results we need to open the log file we have produced.
less SRR2589044_fastq.gz.log
This is cutadapt 3.5 with Python 3.9.5
Command line parameters: -q 20 -a CTGTCTCTTATACACATCT -A CTGTCTCTTATACACATCT -o SRR2589044_1.t
rim.fastq.gz -p SRR2589044_2.trim.fastq.gz SRR2589044_1.fastq.gz SRR2589044_2.fastq.gz
Processing reads on 1 core in paired-end mode ...
Finished in 71.38 s (64 µs/read; 0.93 M reads/minute).
=== Summary ===
Total read pairs processed: 1,107,090
Read 1 with adapter: 175,119 (15.8%)
Read 2 with adapter: 165,406 (14.9%)
Pairs written (passing filters): 1,107,090 (100.0%)
Total basepairs processed: 332,127,000 bp
Read 1: 166,063,500 bp
Read 2: 166,063,500 bp
Quality-trimmed: 27,558,810 bp (8.3%)
Read 1: 5,686,043 bp
Read 2: 21,872,767 bp
Total written (filtered): 290,141,080 bp (87.4%)
Read 1: 152,675,102 bp
Read 2: 137,465,978 bp
Exercise
Use the output from your cutadapt command to answer the following questions.
- How many reads had adapters in the R1 reads?
- What total percent of reads were trimmed/filtered?
Solution
- 15.8%
- 87.4%
We can confirm that we have our output files:
$ ls SRR2589044*
SRR2589044_1.fastq.gz SRR2589044_2.fastq.gz SRR2589044_fastq.gz.log
SRR2589044_1.trim.fastq.gz SRR2589044_2.trim.fastq.gz
The output files are also FASTQ files. It should be smaller than our input file, because we’ve removed reads. We can confirm this:
$ ls SRR2589044* -l -h
-rw-rw-r-- 1 csuser csuser 124M Nov 3 11:05 SRR2589044_1.fastq.gz
-rw-rw-r-- 1 csuser csuser 122M Nov 3 13:22 SRR2589044_1.trim.fastq.gz
-rw-rw-r-- 1 csuser csuser 128M Nov 3 11:05 SRR2589044_2.fastq.gz
-rw-rw-r-- 1 csuser csuser 120M Nov 3 13:22 SRR2589044_2.trim.fastq.gz
-rw-rw-r-- 1 csuser csuser 5.8K Nov 3 13:22 SRR2589044_fastq.gz.log
We’ve just successfully run cutadapt on one of our FASTQ files!
However, there is some bad news. cutadapt can only operate on
one sample at a time and we have more than one sample. The good news
is that we can use a for loop to iterate through our sample files
quickly!
We unzipped one of our files before to work with it, let’s compress it again before we run our for loop.
gzip SRR2584863_1.fastq
$ for infile in SRR2584863 SRR2584866 SRR2589044
> do
> cutadapt -q 20 -a CTGTCTCTTATACACATCT \
> -A CTGTCTCTTATACACATCT \
> -o $infile\_1.trim.fastq.gz -p $infile\_2.trim.fastq.gz \
> $infile\_1.fastq.gz $infile\_2.fastq.gz \
> > $infile\_fastq.gz.log
> done
Go ahead and run the for loop. It should take a few minutes for
cutadapt to run for each of our six input files. Once it’s done
running, take a look at your directory contents. You’ll notice that even though we ran cutadapt on file SRR2589044 before running the for loop, there is only one set of files for it. Because we matched the ending _1.fastq.gz, we re-ran cutadapt on this file, overwriting our first results. That’s ok, but it’s good to be aware that it happened.
$ ls
SRR2584866_1.fastq.gz SRR2589044_1.trim.fastq.gz
SRR2584863_1.fastq.gz SRR2584866_1.trim.fastq.gz
SRR2584863_1.trim.fastq.gz SRR2589044_2.fastq.gz
SRR2584866_2.fastq.gz SRR2589044_2.trim.fastq.gz
SRR2584863_2.fastq.gz SRR2584866_2.trim.fastq.gz
SRR2584863_2.trim.fastq.gz SRR2589044_1.fastq.gz
SRR2584863_fastq.gz.log SRR2584866_fastq.gz.log
SRR2589044_fastq.gz.log
We’ve now completed the trimming and filtering steps of our quality
control process! Before we move on, let’s move our trimmed FASTQ files
to a new subdirectory within our data/ directory.
$ cd ~/cs_course/data/untrimmed_fastq
$ mkdir ../trimmed_fastq
$ mv *.trim* ../trimmed_fastq
$ cd ../trimmed_fastq
$ ls
SRR2584863_1.trim.fastq.gz SRR2584866_1.trim.fastq.gz SRR2589044_1.trim.fastq.gz
SRR2584863_2.trim.fastq.gz SRR2584866_2.trim.fastq.gz SRR2589044_2.trim.fastq.gz
Here is a diagram of what the file structure of your cs_course “data” directory looks like now:
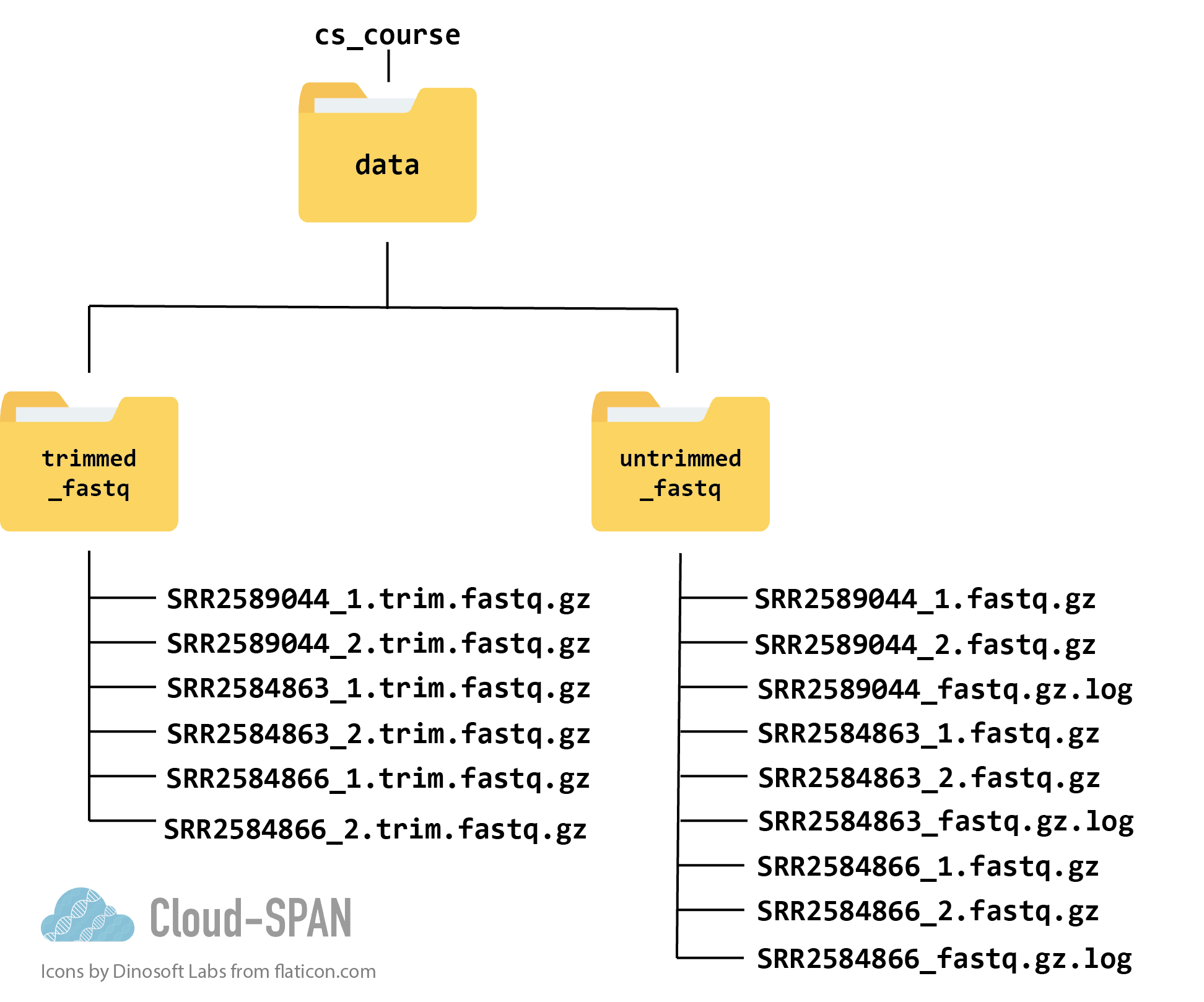
The original data files are still in the untrimmed_fastq folder, while the new trimmed files get new names and reside in the trimmed_fastq directory. There are, of course, other directories containing other information (such as the quality control data we collected last episode).
Bonus Exercise (Advanced)
Now that our samples have gone through quality control, they should perform better on the quality tests run by FastQC. Go ahead and re-run FastQC on your trimmed FASTQ files and visualize the HTML files to see whether your per base sequence quality is higher after trimming.
Solution
In your AWS terminal window do:
$ fastqc ~/cs_course/data/trimmed_fastq/*.fastq*In the terminal on your local machine (not the one logged into the AWS instance) do:
$ mkdir fastqc_html/trimmed $ scp -i login_key_instanceNNN.pem csuser@instanceNNN.cloud-span.aws.york.ac.uk:/home/csuser/cs_course/data/trimmed_fastq/*.html fastqc_html/trimmedThen take a look at the html files in your browser.
Remember to replace everything between the
@and:in your scp command with your AWS instance number.After trimming and filtering, our overall quality is much higher, we have a distribution of sequence lengths, and more samples pass adapter content. However, quality trimming is not perfect, and some programs are better at removing some sequences than others. Because our sequences still contain 3’ adapters, it could be important to explore other trimming tools like Trimmomatic to remove these, depending on your downstream application. Cutadapt did pretty well though, and its performance is good enough for our workflow.
Key Points
The options you set for the command-line tools you use are important!
Data cleaning is an essential step in a genomics workflow.